The Big Picture
To watch a chemical reaction unfold, you need a way to trigger it and a way to probe what happens next. We do this with pairs of ultrashort laser pulses. The first initiates the chemistry; the second interrogates the molecule at a precisely controlled time delay. Repeating this measurement across a range of delays builds up a direct picture of how the system evolves from the first moment of excitation through to the final products.
Different probes reveal different aspects of the same underlying dynamics. Mass spectrometry tracks the appearance and disappearance of reaction products. Photoelectron and photoion velocity-map imaging give access to the momentum distributions of the fragments. Coulomb explosion imaging reconstructs the three-dimensional nuclear geometry directly. Together, these techniques let us follow a reaction across multiple observables simultaneously, providing rich experimental datasets that benchmark quantum chemical theory and molecular dynamics simulation.
Our ambition is to push these capabilities beyond their current limits; studying bimolecular reactions, electron-driven chemistry, and thermally-activated dynamics that have so far been largely out of reach.
CURRENT PROJECTS
-
Ultrafast Dissociative Ionisation
When a molecule is ionised, the conventional wisdom is that the internal energy equilibrates throughout the molecule before any reaction occurs. This statistical picture underpins most of mass spectrometry, but it breaks down when reactions are faster than intramolecular vibrational redistribution. Understanding when and why this happens — and what it means for reaction outcomes — is an open problem with direct relevance to how we model and predict ionisation rich environments, e.g. radiation chemistry and industrial plasmas, as well as how we can simulate mass spectrometry outcomes.
We use strong-field ionisation with a femtosecond laser pulse to prepare molecular ions in an ensemble of excited states, then probe the subsequent fragmentation with a second pulse using time-resolved mass spectrometry. This lets us follow multiple reaction channels simultaneously, tracking the connections between reactants, intermediates, and products in a single measurement. We complement these experiments with Born-Oppenheimer molecular dynamics simulations developed in-house, which allow us to directly compare experimental observables with theoretical predictions of the evolving ion chemistry.
We are extending this work to larger, non-volatile molecules using a laser desorption source to access the fundamental insights from gas phase studies for larger, currently out-of-reach molecules.

Electron ionisation of DNA, migration of the hole along the backbone, and finally strand breakage.
-
Coulomb Explosion Imaging
Coulomb explosion imaging (CEI) is our most powerful structural probe. A short, intense laser pulse strips multiple electrons from a molecule faster than the nuclei can respond, causing it to explode. Measuring the momenta of the resulting fragment ions in coincidence reconstructs the three-dimensional nuclear geometry at the moment of explosion. Repeated at a series of pump-probe time delays, this builds up a direct picture of the nuclear wavepacket as it evolves during a reaction.
The combination of spatial resolution, three-dimensional sensitivity, and femtosecond time resolution that CEI offers is not matched by any other technique. Performing these experiments at the high pulse energies available at X-ray Free Electron Laser facilities unlocks the full potential of the method. We work at the Small Quantum Systems instrument at the European XFEL, where we have been studying the UV-induced photochemistry of cyclic disulfides — biologically relevant functional groups whose dynamics are a stringent test of quantum molecular dynamics theory. Our results show that state-of-the-art simulations of these systems are quantitatively inaccurate, a finding that is only possible because CEI gives us direct access to the nuclear geometry rather than an indirect spectroscopic proxy.
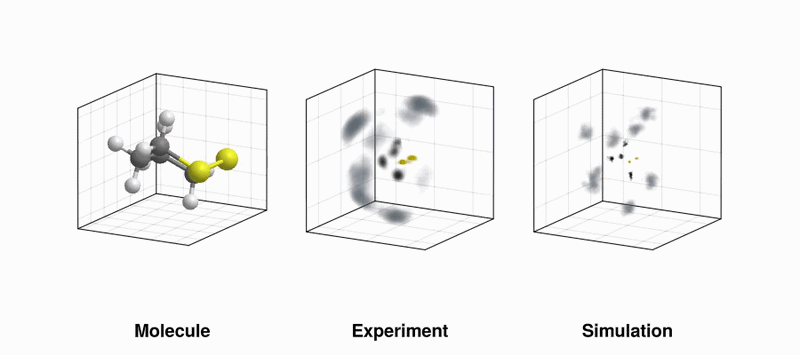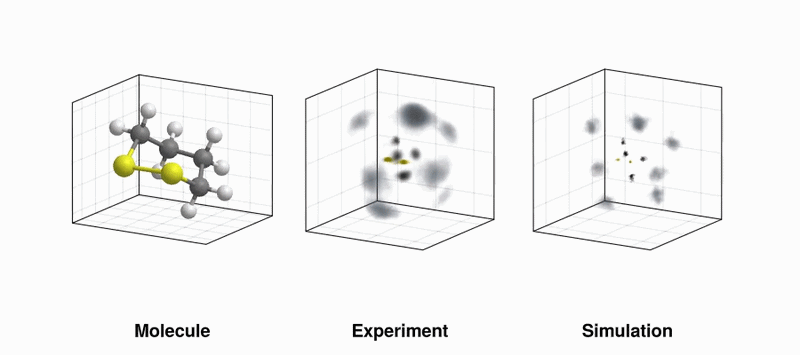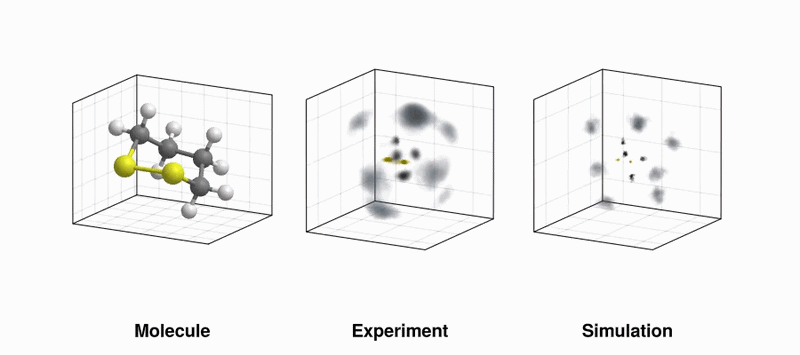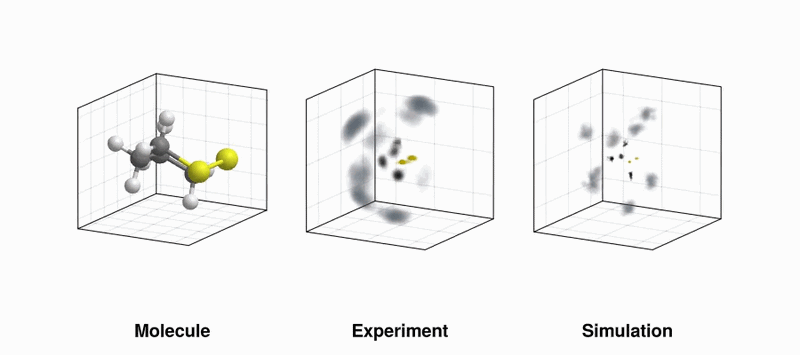
(LEFT) Our favourite molecule, 1,2-dithiane; (MIDDLE) Experimentally measured velocity vector distribution in the laboratory frame of sulfur (yellow), carbon (black), and hydrogen (grey) ions measured by Coulomb explosion; (RIGHT) Same as in the middle panel, but using simulated data (Thanks to Benoit Richard at EuXFEL for the gif!)
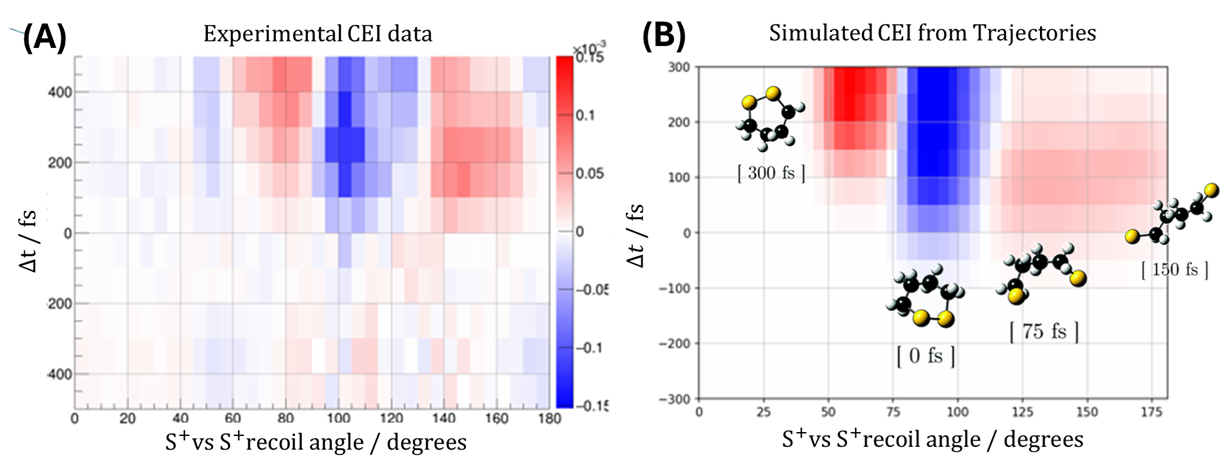
UV-induced structural dynamics of 1,2-dithiane. (LEFT) Recoil frame covariance maps of sulfur ions recoiling from another sulfur ion travelling in the positive x-direction at the same time steps; (RIGHT) Same observable but using simulated data with the evolving molecular structure taken from quantum dynamics simulations superimposed
-
Time-Resolved Photoelectron and Photoion Imaging
Velocity-map imaging (VMI) records the full momentum distribution of photoelectrons or photoions ejected from a molecule by a probe laser pulse. Combined with a pump-probe scheme, this gives access to how the electronic and nuclear structure of a molecule evolves in time. Between TOF-MS, CEI, and VMI, we get a full experimental description of the time-resolved dynamics of our system of interest.
Right now, we are starting to use time-resolved photoelectron imaging to study endohedral fullerenes — molecules in which an atom or small molecule is encapsulated inside a carbon cage. These systems are proposed as candidates for quantum information science applications, where the cage acts as a shield that isolates the guest from its environment. A key open question is how strongly does the guest interact with the cage, and what are the mechanism of loss of coherence? By probing the photoelectron dynamics of the encapsulated species, we aim to quantify these host-guest interactions directly.
A related direction we are developing is the use of time-resolved ion and photoelectron imaging to study rotational wavepacket dynamics in photodissociation. Preparing a coherent superposition of rotational states and watching it evolve through alignment, dephasing, and rephasing gives access to interference effects in isolated molecules. Understanding these quantum coherence effects in the gas phase is a natural stepping stone towards the same questions in the endohedral context.
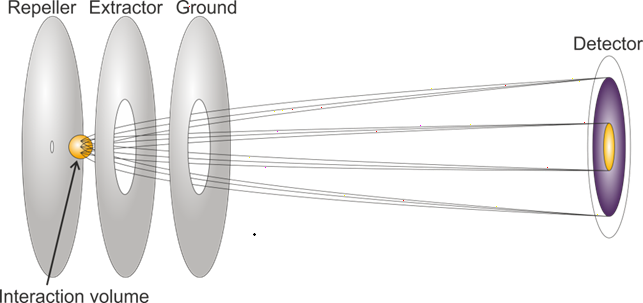
Schematic representation of a velocity map imaging spectrometer